Садржај
- Узроци
- Симптоми
- Испити и тестови
- Третман
- Групе за подршку
- Када контактирати медицинског стручњака
- Превенција
- Алтернативе Намес
- Имагес
- Референце
- Датум прегледа 8/6/2017
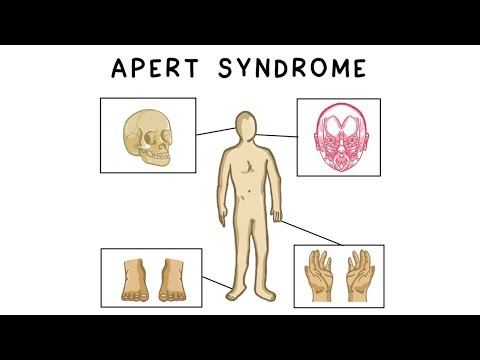
Апертни синдром је генетска болест у којој се шавови између костију лобање затварају раније него што је нормално. То утиче на облик главе и лица.
Узроци
Апертни синдром се може преносити кроз породице (наслеђене) као аутосомно доминантно својство. То значи да само један родитељ треба да пренесе неисправан ген да би дете имало стање.
Неки случајеви се могу појавити без познате породичне историје.
Апертни синдром је узрокован једном од двије промјене ФГФР2 гене. Овај дефект гена узрокује да се неки од коштаних шавова лобање затвори прерано. Ово стање се назива краниосиностоза.
Симптоми
Симптоми укључују:
- Рано затварање шавова између костију лобање, забиљежено код сечења дуж шавова (краниосиностоза)
- Честе инфекције уха
- Фузија или јака трака 2., 3. и 4. прста, често названа "рукавицама"
- Губитак слуха
- Велика или касна мека мрља на бебиној лобањи
- Могући, спор интелектуални развој (варира од особе до особе)
- Видљиве или испупчене очи
- Јака неразвијеност средњег слоја
- Абнормалности скелета (екстремитета)
- Низак
- Ткање или спајање прстију
Неколико других синдрома може довести до сличног изгледа лица и главе, али не укључују тешке особине Аперт синдрома. Ови слични синдроми укључују:
- Царпентер синдром (клееблаттсцхадел, деформација лобање цловерлеаф)
- Цроузонова болест (краниофацијална дисостоза)
- Пфеиффер синдром
- Саетхре-Цхотзенов синдром
Испити и тестови
Здравствени радник ће обавити физички преглед. Рендген за руке, стопала и лобање ће бити обављен. Тестове слуха треба увек изводити.
Генетичко тестирање може потврдити дијагнозу Аперт синдрома.
Третман
Лечење се састоји од операције за исправљање абнормалног раста костију лобање, као и за фузију прстију и прстију. Деца са овим поремећајем треба прегледати специјализовани тим за краниофацијалну хирургију у дечјем медицинском центру.
Ако има проблема са слухом, треба консултовати специјалисте за слух.
Групе за подршку
Дјечје краниофацијално удружење: ввв.ццакидс.цом
Када контактирати медицинског стручњака
Позовите свог провајдера ако имате породичну историју Аперт синдрома или приметите да се лубања ваше бебе не развија нормално.
Превенција
Генетско саветовање може бити од помоћи ако имате породичну историју овог поремећаја и планирате да затрудните. Ваш лекар може тестирати бебу на ову болест током трудноће.
Алтернативе Намес
Ацроцепхалосиндацтили
Имагес

Синдацтили
Референце
Кинсман СЛ, Јохнстон МВ. Конгениталне аномалије централног нервног система. У: Клиегман РМ, Стантон БФ, Ст. ЈВ, Сцхор НФ, едс. Нелсон Тектбоок оф Педиатрицс. 20тх ед. Пхиладелпхиа, ПА: Елсевиер; 2016: цхап 591.
Робин НХ, Фалк МЈ, Халдеман-Енглерт ЦР. Синдроми краниосиностоза везани за ФГФР. ГенеРевиевс. 2011: 11. ПМИД: 20301628 ввв.нцби.нлм.них.гов/пубмед/20301628.
Датум прегледа 8/6/2017
Ажурирано: Анна Ц. Еденс Хурст, МД, МС, доцент медицинске генетике, Универзитет Алабама у Бирмингему, Бирмингем, АЛ. Преглед је пружила ВериМед Хеалтхцаре Нетворк. Прегледали су га и Давид Зиеве, МД, МХА, медицински директор, Бренда Цонаваи, уредница редакције, и А.Д.А.М. Уреднички тим.